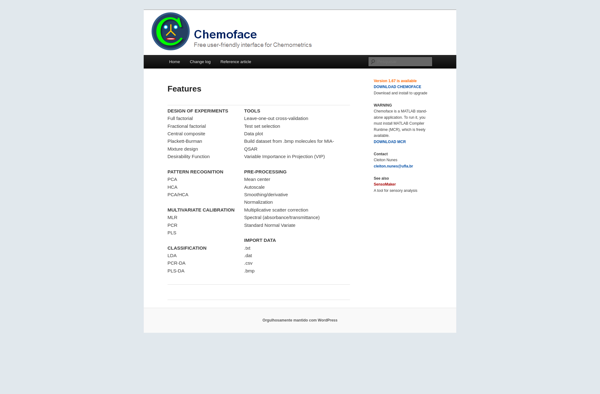
Chemoface
Description: Chemoface is open-source software for predicting the biological activities of small molecules based on their chemical structures. It uses machine learning models trained on datasets of compounds and their bioactivities.
Type: software
Pricing: Open Source
SAS JMP
Description: SAS JMP is a comprehensive statistical analysis and data visualization software used by statisticians, engineers, scientists, quants, and other data analysts. It provides interactive graphics, predictive modeling, and data analysis capabilities for statistical analysis and data mining.
Type: software