Chemoface is open-source software for predicting the biological activities of small molecules based on their chemical structures. It uses machine learning models trained on datasets of compounds and their bioactivities.
Chemoface: Open-Source Software for Predicting Biological Activities
Open-source software for predicting biological activities of small molecules based on chemical structures, using machine learning models trained on compound datasets.
What is Chemoface?
Chemoface is an open-source computer program for predicting the biological activities of chemical compounds. It utilizes machine learning models that have been trained on large datasets of chemicals and their associated bioassay data to predict potential therapeutic effects and safety risks.
The key capabilities of Chemoface include:
Predicting activity against a range of drug targets like enzymes, ion channels, and protein-protein interactions
Estimating absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters
Screening libraries of compounds to identify structures with desired bioactivities
Prioritizing compounds for synthesis and testing
Understanding structure-activity relationships
Some of the machine learning methods used by Chemoface include neural networks, random forests, support vector machines, and graph convolutions. It can make predictions based on simplified molecular-input line-entry system (SMILES) representations of compound structures.
Chemoface is implemented in Python and has an open-source MIT license allowing free usage. It provides both command line and graphical user interfaces. The models are customizable and new training data can be added to tailor predictions to specific drug discovery projects.
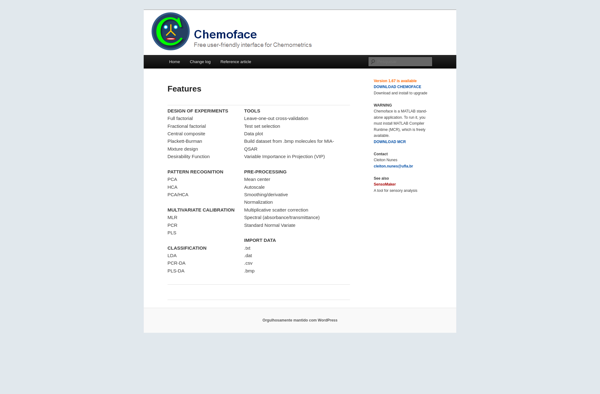
Chemoface Features
Features
Predict biological activities of small molecules
Uses machine learning models trained on bioactivity datasets
Open-source software
Web-based graphical user interface
Support for multiple machine learning algorithms
Built-in datasets of compounds and bioactivities
Custom model training
Activity predictions and statistical analysis
2D and 3D molecular structure visualization
Structure-based virtual screening
Pricing
Open Source
Pros
Free and open-source
User-friendly interface
Pre-trained models available
Customizable model building
Supports major machine learning methods
Can handle large datasets
Visualization capabilities
Active development and community
Cons
Requires machine learning expertise for full utilization
R is an open-source programming language and free software environment for statistical computing, bioinformatics, graphics, data science, and general-purpose programming. The R language provides a wide variety of statistical analysis techniques and graphical capabilities which make it a popular choice for data analysis and visualization.Some key features of R include:Open-source...
Minitab is a comprehensive software package used for statistical analysis and quality improvement applications. It has a wide range of analytical capabilities including basic statistics, regression analysis, design of experiments, statistical process control charts, reliability analysis, and more.Some key features and benefits of Minitab:User-friendly spreadsheet-style interface for entering, viewing, and...
RStudio is a popular open-source IDE for R, a programming language for statistical computing and graphics. It provides a user-friendly graphical user interface that makes working with R much easier by integrating tools for plotting, debugging, workspace management, and other features.Some key features of RStudio include:Code editor with syntax highlighting,...
Deducer is a user-friendly open-source data analysis software aimed at beginners looking to learn statistics and data analysis using R without having to code. It was designed by academic researchers to be a free easy-to-use alternative to proprietary statistical software packages.Deducer provides a graphical user interface to R that allows...
RKWard is a free and open-source integrated development environment for the R statistical programming language. It provides a graphical user interface that allows users to work with R without needing to manually type code.Some key features of RKWard include:Code editor with syntax highlighting, auto-completion and other productivity toolsData viewer to...
DOE++ is an open-source, extensible software framework designed to streamline and automate various aspects of the scientific research process. Some key features and capabilities of DOE++ include:Intuitive graphical user interface for designing experiments, including full factorial, fractional factorial, response surface, mixture, and custom designs.Seamless integration of data analysis tools like...
PSPP is a free and open source alternative to the proprietary IBM SPSS Statistics software. It aims to provide similar capabilities and features for statistical analysis, data management, and graphics as SPSS Statistics does, while being available at no cost under the GNU General Public License.Some key features and capabilities...
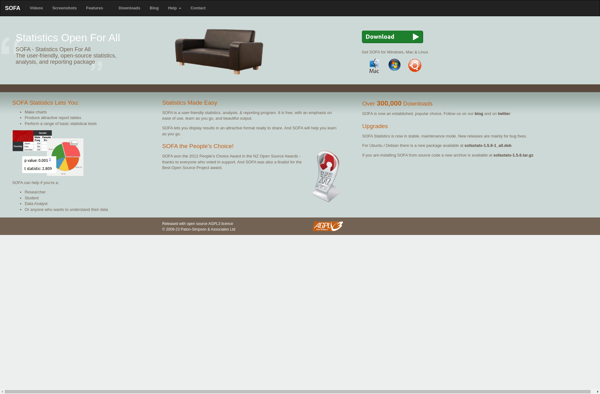
SOFA Statistics is a user-friendly, open-source statistics, analysis, and reporting tool that runs as a desktop application on Windows, Mac, and Linux. It provides an extensive range of statistical methods and visualization tools for exploratory data analysis, data cleaning, model fitting, results reporting, and dashboard creation.Some key features of SOFA...
SAS JMP is a comprehensive statistical analysis and data visualization software application developed by SAS Institute. It provides a visual and interactive platform for data analysis, enabling users to analyze data, build statistical and predictive models, and generate custom reports.Some key features and capabilities of JMP include:Interactive and dynamic graphs...
The R Commander is a basic-statistics graphical user interface for R, an open source programming language and software environment for statistical analysis, data manipulation, and graphics visualization. The R Commander provides a simple way to utilize many R features through menus, dialog boxes, and other user interface controls rather than...
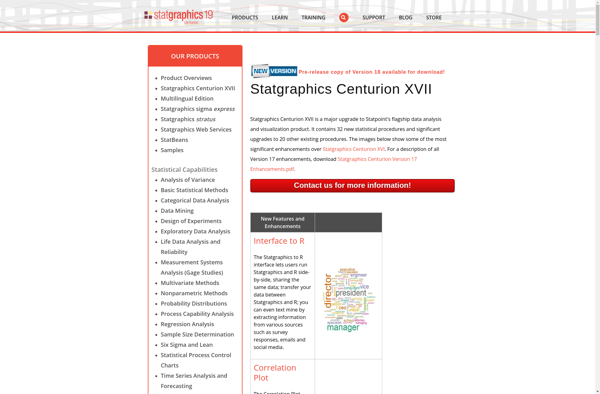
Statgraphics Centurion XVII is a comprehensive statistical data analysis and visualization software used by businesses, research organizations, quality improvement teams, and others to make data-driven decisions.It provides over 250 statistical procedures including analysis of variance, regression analysis, design of experiments, process capability analysis, multivariate data analysis, nonparametric tests, and more....
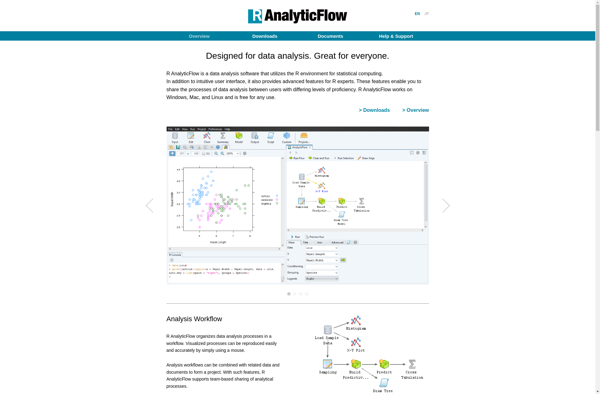
R AnalyticFlow is an open-source data analysis platform built specifically for the R programming language. It allows data scientists and analysts to create reusable analysis flows that connect various data sources, R scripts, and visualization code together into an end-to-end pipeline.Some key features of R AnalyticFlow include:Visual workflow editor -...
PAST is a comprehensive, user-friendly software package for scientific data analysis, with a focus on paleontology and paleobiology research. Developed by researchers at the University of Oslo, PAST provides a wide range of analytical tools and statistical tests within an intuitive graphical user interface.Key features of PAST include:Data manipulation -...
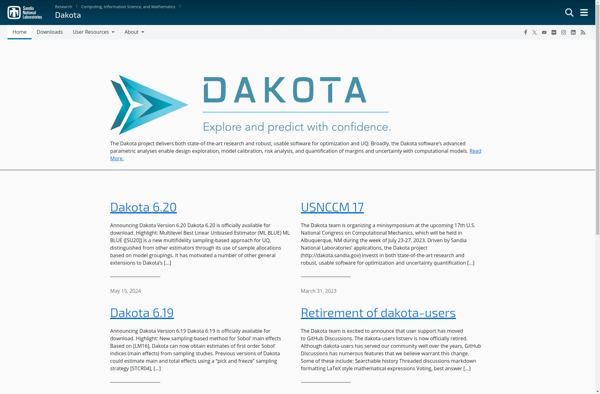
Dakota (Design Analysis Kit for Optimization and Terascale Applications) is an extensible open-source software for design optimization, parameter estimation, uncertainty quantification, and sensitivity analysis. It interfaces with simulation codes written in C, C++, Fortran, Python, and MATLAB.Some key capabilities of Dakota include:Optimization with gradient and non-gradient-based methods, heuristic techniques, hybrid...
Revolution Analytics' Revolution R is a software environment and suite of tools designed to provide an enhanced user experience for the R programming language. Some key features of Revolution R include:Integrated Development Environment (IDE) - Provides an interface for authoring, debugging, and executing R code, with features like syntax highlighting,...
SOSstat is an open-source statistical analysis program designed specifically for educational purposes in teaching statistics. It provides an easy-to-use interface allowing students to conduct basic statistical analyses and graphing without needing extensive statistical knowledge.Key features of SOSstat include:Conducting common statistical tests like t-tests, ANOVA, correlation, regression, and nonparametric testsGenerating graphs...